Human Kidney meQTL Atlas
Methylation quantitative trait loci of human kidney
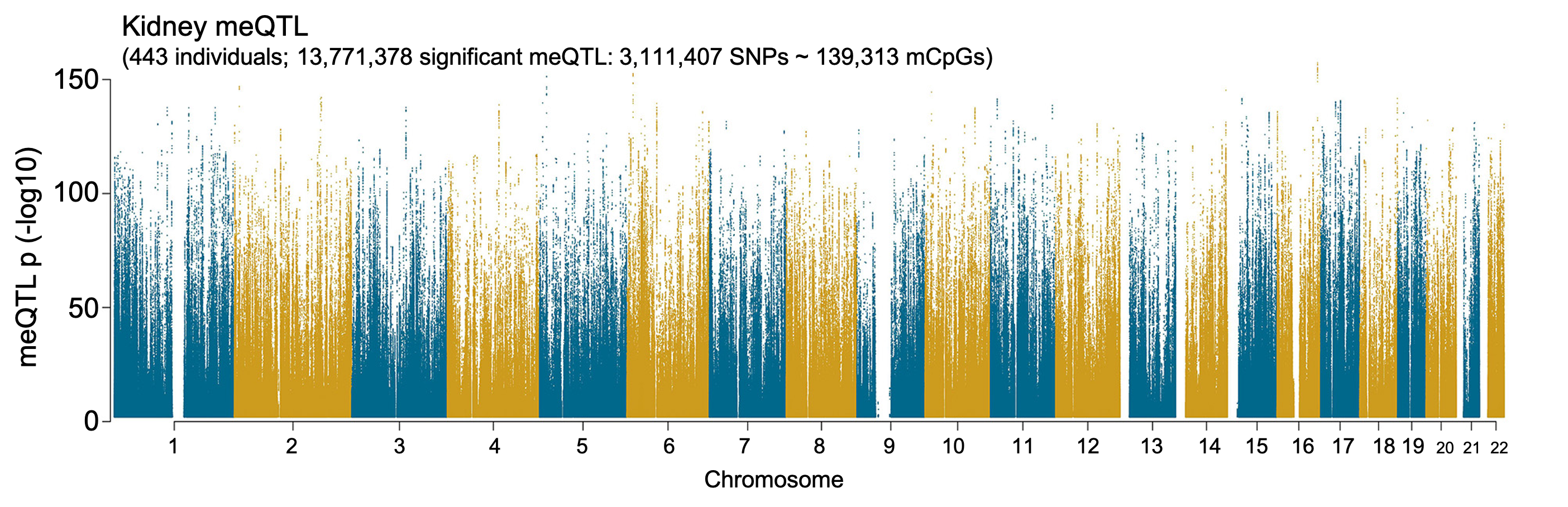
The epigenome describes the cellular gene expression regulatory logic and integrates effects from genetic variation and environmental changes. Thus, epigenomic profiling data could provide critical insight into kidney disease development, as they have in previous work for other complex diseases. DNA methylation, a key component of the epigenome, regulates gene expression by altering transcription factor binding strength or recruiting proteins involved in gene repression. Here we conducted cis-meQTL association analysis using 443 samples with imputed genotyping data and methylation data by EPIC array, and defined the genetic effects on DNA methylation. Our analysis indicates that kidney meQTLs mediate a higher fraction of heritability for kidney function traits (eGFRcrea, eGFRsys and BUN) using individual-level methylation profiles from the same human kidney samples, and highlights that new epigenetic datasets will be critical for GWAS functionalization. To explore the relationship between cytosine methylation and gene expression, we obtained gene expression information via RNA sequencing for the analyzed kidney samples (N=414) and performed expression quantitative trait methylation (eQTM) analysis. The integration of meQTL and eQTM provides a new way to prioritize target genes for eGFRcrea GWAS variants.